Introduction
Only mRNA containing 7-methyl guanosine (M7G) cap structure at the 5'end and a Poly(A) tail at the 3' end can be effectively expressed in eukaryotic cells.Capped RNA synthesis kit enables the production of a large number of RNA containing a 5' Cap structure in vitro, which is capped to the 5' end of RNA by the use of anti-reverse Cap Analog (ARCA).
ARCA structure: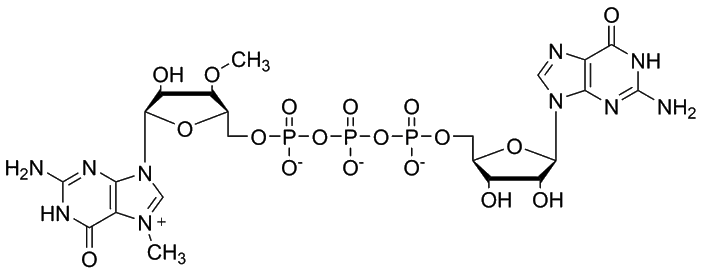
Using the control template supplied with the kit, each reaction will yield approximately 60-70μg of RNA.
Poly(A) tailing of RNA
Tailings of mRNA can be obtained by Poly(A) Polymerase. Using this kit, approximately 100 base poly(A) tails can be added to RNA transcripts.
Materials provided with the kit:
The kit contains sufficient reagents for 10 reaction of 20μL each. Each standard reaction yield up to 70μg of capped RNA, 2300nt length, from 1 μg control template.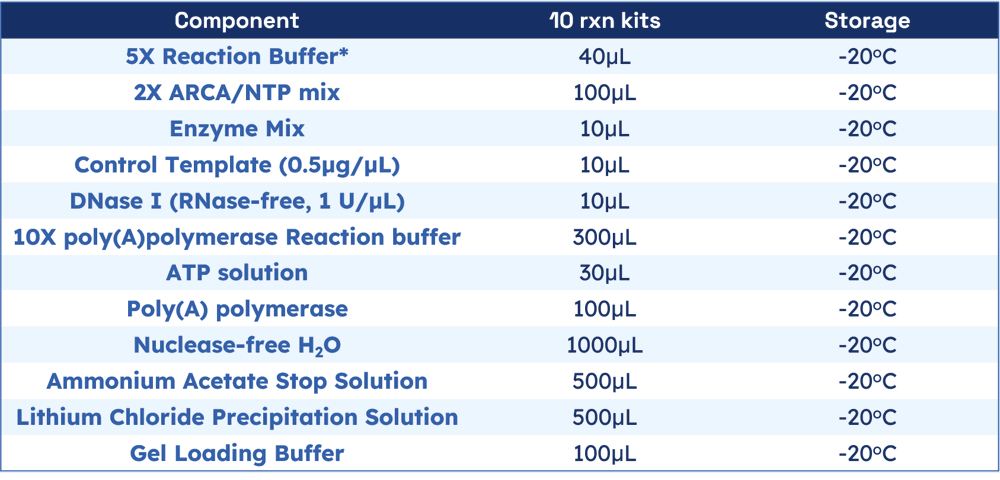
* Salts, buffer, dithiothreitol, and other ingredients; 5X Reaction Buffer store at -70°C may result in the formation of a white precipitate. If this happens, heat the tube to 37°C for 5 minutes and mix thoroughly to resuspend the precipitate.
Kit procedure
Preparation of template DNA
Linearized plasmid DNA and PCR products that contain a T7 polymerase promoter site can be used as templates for in vitro transcription with this kit. In general, any DNA with a T7 promoter site that is pure enough to be easily digested with restriction enzymes can be used for in vitro transcription.
Figure 1 T7 polymerase promoter: Minimal Sequence Requirements
T7 +1
TAATACGACTCACTATAGGGAGA
The +1 base (in bold) is the first base incorporated in RNA during transcription. The underline shows the minimum promoter sequence needed for efficient transcription.
Plasmid templates
DNA templates are usually linearized prior to in vitro transcription to produce RNA of defined length. Linearize the DNA by digestion with an appropriate restriction endonuclease followed by an appropriate clean-up procedure, such as phenol extraction followed by ethanol precipitation. Start with at least 30% more DNA than is required for the transcription reaction to allow for DNA loss during purification and visualization by gel electrophoresis.
The restriction enzyme used to linearize the plasmid should leave blunt ends or 5' overhangs (linearization of template with an enzyme that produces a 3' overhang will result in aberrant transcripts). DNA should be digested in a minimum volume of 10μL per ug of DNA, using the buffer and temperature recommended for each enzyme.
Linearized DNA can be used straight from the digestion reaction if desired, with only a slight reduction of yield (< 10%). However, digestion reactions must be heat-denatured at 65°C for 20 minutes prior to transcription. Template linearized DNA from restriction digestion reaction should not constitute more than 10% of the total transcription volume.
Optimal RNA yields depend on a high-quality DNA template. The DNA template must be free of RNase. If the presence of RNase is suspected, treat the DNA with ProteinaseK (100μg/mL) and SDS (0.5%) in 50 mM Tris-HCl (pH7.5), 5mM CaCl2 for 30minutes at 37°C. Purify the DNA further by extraction with TE-saturated (pH8.0) phenol:chloroform: isopropanol (25:24:1) and ethanol precipitation.
PCR Templates
DNA generated by PCR can be transcribed directly from the PCR provided it contains a T7 Polymerase promoter upstream of the sequence to be transcribed. PCR products should be examined on an agarose gel before use as a template in to estimate concentration, and to verify that the products are unique and the expected size.
Figure 2 PCR prime example
T7 +1
5'-TAATACGACTCACTATAGGGtgaattgcgcaatactgactgg-3'
The +1 base (in bold) is the first base incorporated in RNA during transcription. The underline shows the minimum promoter sequence needed for efficient transcription. The lower-case sequence is gene-specific sequence.
mRNA Synthesis Protocols
We strongly recommend wearing gloves and using nuclease-free tubes and reagents to avoid RNase contamination. Reactions are typically 20μL but can be scaled up and down as needed. Reactions should be assembled in nuclease-free microfuge tubes or PCR strip tubes.
1. Assemble transcription reaction at room temp.
The spermidine in the 5X Reaction Buffer can coprecipitate the template DNA if the reaction is assembled on ice.
Add the 5X Reaction Buffer after the water and the ribonucleotides are already in the tube.
The following amounts are for a single 20μL reaction. Reactions may be scaled up or down if desired.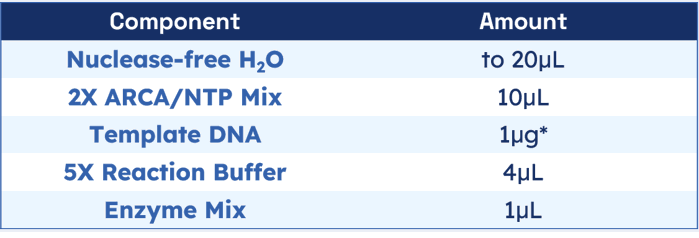
*The transcript may vary depending on the amount of template DNA.
2. Mix thoroughly
Gently flick the tube or pipette the mixture up and down gently, and then microfuge tube briefly to collect the reaction mixture at the bottom of the tube.
3. Incubate at 37°C, 2 hr.
Typically, 2 hours of incubation is sufficient. Incubation time can be extended to 4 hours for maximum yield.
4. (optional) Add 1μL DNase (RNase-free), mix well and incubate 30 min at 37°C.
This DNase treatment removes the template DNA. For many applications it may not be necessary because the template DNA will be present at a very low concentration relative to the RNA.
a. Add 1μL DNaseI,and mix well (the reaction may be viscous).
b. Incubate at 37°C for 30min.
5. Poly(A) tailing procedure. Set up the tailing reaction as below. The IVT reaction solution does not need to be purified.
Mix thoroughly and pulse-spin in a microfuge. Incubate at 37°C for 30 minutes.
30min tailings will provide A poly(A) tail of 150nt or longer for most mRNAs. If a longer polyA tail is needed, the time can be extended to 45min.
6.Analysis of transcription products by gel electrophoresis.
a. To get good resolution of the RNA, load 0.2-1μg per gel lane.
b. To each sample, add three volumes of Gel Loading Buffer.
c. Heat for 3-5min at 70°C.
d. Load onto gel.
e. Visualizing RNA by staining the gel with ethidium bromide.
Recovery of the RNA
The degree of purification required after the transcription reaction depends on what will be done with the RNA. Two different methods follow; choose one or more according to your application and resources.
Ammonium acetate precipitation
Selectively precipitates RNA, while leaving most of the protein and unincorporated rNTP in the supernatant. Note: for this method, the RNA to be purified must be >100 bases in size.
1) Add one volume of 5 M ammonium acetate (21μL for the standard reaction), mix well.
2) Incubate for 30 minutes on ice.
3) Pellet the RNA by centrifugation at >10,000 x g for 15 minutes at 4°C.
4) Remove the supernatant with a pipette and gently rinse the pellet with 70% ethanol.
5) Remove the 70% ethanol with a pipette without disturbing the RNA pellet.
6) Allow pellet to dry, then resuspend in Nuclease-free Water, TE or other suitable buffer.
7) While usually unnecessary, steps 1-6 may be repeated a second time for even cleaner RNA.
8) Allow the pellet to dry, then resuspend in 25μL of Nuclease-free Water or TE buffer
9) Store frozen at -20°C or -70°C.
Lithium chlorideprecipitation
Lithium Chloride (LiCl) precipitation is a convenient and effective way to remove unincorporated nucleotides and most proteins. Lithium chloride precipitation, however, does not precipitate transfer RNA and may not efficiently precipitate RNAs smaller than 300 nucleotides. Also, the concentration of RNA should be at least 0.1 μg/μL to assure efficient precipitation. To precipitate from reactions that are thought to have very low yields of RNA, do not dilute the transcription reaction with water prior to adding the LiCl Precipitation Solution in the step below.
1) Stop the reaction and precipitate the RNA by adding 30μL Nuclease-free Water and 30μL LiCl Precipitation Solution.
2) Mix thoroughly. Incubate for 30 minutes on ice.
3) Pellet the RNA by centrifugation at >10,000 x g for 15 minutes at 4°C.
4) Remove the supernatant with a pipette and gently rinse the pellet with 70% ethanol.
5) Remove the 70% ethanol with a pipette without disturbing the RNA pellet.
6) Allow pellet to dry, then resuspend in Nuclease-free Water, TE or other suitable buffer.
7) While usually unnecessary, steps 1-6 may be repeated a second time for even cleaner RNA.
8) Allow the pellet to dry, then resuspend in 25μL of Nuclease-free Water or TE buffer.
9) Store frozen at -20°C or -70°C.
Phenol: chloroform extraction and isopropanol precipitation
It will remove all enzyme and most of the free nucleotides from High Yield T7 RNA Synthesis Kit reactions.
1) Add 115μL Nuclease-free Water and 15μL 5M ammonium acetate, and mix thoroughly.
2) Extract with an equal volume of phenol/chloroform (it can be water-saturated, buffer-saturated, or acidic), and then with an equal volume of chloroform. Recover aqueous phase and transfer to new tube.
3) Precipitate the RNA by adding 1 volume of isopropanol and mixing well.
4) Incubate for 15 minutes at -20°C.
5) Pellet the RNA by centrifugation at >10,000 x g for 15 minutes at 4°C.
6) Remove the supernatant with a pipette and gently rinse the pellet with 70% ethanol.
7) Remove the 70% ethanol with a pipette without disturbing the RNA pellet.
8) Allow pellet to dry, then resuspend in Nuclease-free Water, TE or other suitable buffer.
9) Allow the pellet to dry, then resuspend in 25μL of Nuclease-free Water or TE buffer.
10) Store frozen at -20°C or -70°C.
Quantitating RNA Yield Using Spectrophotometry
Reading the A260 of a diluted aliquot of the reaction is clearly the simplest way to determine yield, but any unincorporated nucleotides and/or template DNA in the mixture will contribute to the reading. Note Accurate spectrophotometric measurement requires an OD260 ≥ 0.05.
1. Blank the spectrophotometer at 260 nm with an appropriate buffer (e.g., 10mM Tris, pH7.5) near neutral pH.
2. Prepare an appropriate dilution of the RNA (1:50–1:100) in the same buffer used to blank the spectrophotometer. Place a piece of laboratory film (e.g., Parafilm® laboratory film) over the top of the cuvette and mix the sample well. The conversion factor for RNA is 0.040μg/μL per OD260 unit. Take the spectrophotometric reading. For a reading of 0.50, calculate the concentration as follows:
Concentration = A260 × dilution factor × conversion factor
Example 0.50 × 500/5 × 0.040μg/μL = 2μg/μL
3. Calculate the yield of RNA by multiplying the volume in microliters by the concentration. The sample volume in this example is 25μL, therefore, the RNA yield is 50μg.
